|
- What is the single letter code for amino acids?
- How do I look in the RESID Database for all the known modifications of a particular amino acid?
- How do I find the UniProt Knowledgebase entries containing a particular modification annotated in the RESID Database? And how would I find which RESID Database entry corresponds to a modification annotated in the feature table of a UniProt Knowledgebase entry?
- In many RESID Database entries, there are two blocks, a “Formula Block” and a “Correction Block”. What is the difference between these two blocks?
- How are the Formula Block and Correction Block used?
- Why are there “chemical” and “physical” weights? What is the difference?
- How do I look in the RESID Database for all the known modifications with a particular mass difference?
- How are the entries in the RESID Database produced, and how frequently is the RESID Database updated?
- How do I search the RESID Database for entries that were created or changed before or after a particular date?
- Where is the “controlled vocabulary” for the RESID Database?
- The formulas and masses do not seem to correspond to the amino acids. How are the formulas and masses calculated, and how should they be used?
- If the process of methylation replaces a hydrogen with a methyl group giving a net change of (C 1 H 2) or +14.015650 Da, why is the difference formula for N,N-dimethylproline (C 2 H 5), and why are the difference formulas for N,N,N-trimethylalanine and N6,N6,N6-trimethyllysine given as (C 3 H 7), while the difference formula for Nω,Nω,Nω’-trimethylarginine is (C 3 H 6)?
|
Q: What is the single letter code for amino acids?
A: The one-letter and the three-letter codes, which have been used to represent polypeptide sequences since the mid 1960's, were defined by the IUPAC and IUBMB in 1983 as shown in this table.
|
N-formylmethionine, one of the three special encoded amino acids, is translated
for the initiation codon in prokaryotes and in eukaryotic organelles. It is
abbreviated "fMet", but uses the single letter code "M" and is not assigned its
own single letter code. In 2008 the UniProt Knowledgebase began representing encoded selenocysteine and pyrrolysine residues by the IUBMB single letter codes "U" and "O", accompanied by the new feature descriptions
as appropriate. That UniProt Knowledgebase announcement may be viewed here
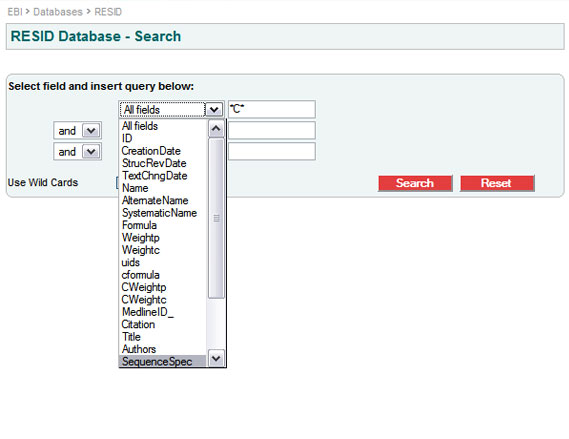
Q: How do I look in the RESID Database for all the known modifications of a particular amino acid?
A: Let us look for the modifications of cysteine, the amino acid with the most different kinds of modifications. Using the SRS search engine, either through http://www.ebi.ac.uk/resid/search.html or through SRS directly, pull down and select “SequenceSpec” in the selection box that shows “All fields” or “AllText”. Then in the adjacent text entry box type “C”, the single letter code for cysteine, and finally hit the "Search" button.
This gives the modifications that can arise from cysteine by itself. In order to see the modifications and cross-links that can arise from cysteine with some other amino acid you need to use wild-cards; type “*C*”.
In order to list only the modifications of cysteine with something else, type “*C*” in the first field selecting SequenceSpec, then combine that with a second field using a “not” or “but not” operator, and put “C” in the second field also selecting “SequenceSpec”.
The entry for cystine, RESID:AA0025, comes out in the list for “C”, cysteine alone, because cystine can arise either from two encoded cysteines, or from a single encoded cysteine forming a disulfide bond with a free (unencoded) cysteine. The two “SequenceSpec” records for RESID:AA0025 are “C, C” and “C”.
|
Q: How do I find the UniProt Knowledgebase entries containing a particular modification annotated in the RESID Database? And how would I find which RESID Database entry corresponds to a modification annotated in the feature table of a UniProt Knowledgebase entry?
A: Because we try to annotate the protein modifications in the UniProt Knowledgebase with feature descriptions that are unique and consistent with a controlled vocabulary, in almost all cases the feature descriptions can themselves serve as identifiers, they do not require a numeric, non-descriptive identifier. (They are what bioinformaticians refer to as “foreign keys”.) Within the XML records of the RESID Database there are database search keys constructed from the feature “FT Key”, the standard part of the feature description, and the amino acid code for the corresponding location in the sequence of a UniProt Knowledgebase entry. If you use the SRS search engine at the EBI, these keys are used dynamically in the “ResidEntry” view of a RESID Database entry to hot-link the features to a search for the corresponding features in UniProt Knowledgebase entries. Likewise, in the “SwissEntry” and the “Related Data” views of a UniProt Knowledgebase entry, the features are hot-linked to the corresponding modifications annotated in the RESID Database.
|
Q: In many RESID Database entries, there are two blocks, a “Formula Block” and a “Correction Block”. What is the difference between these two blocks?
A:The formula block presents the formula and calculated masses for the complete entity, consisting of the amino acid residues with whatever else is gained or lost in the modification. An amino acid “residue” is that part of an amino acid as it is found in a protein, the free amino acid without a hydrogen of the alpha-amino or alpha-imino group and without the hydroxyl of the alpha-carboxyl group.
The correction formula then presents only whatever is gained or lost in the modification, and is always based on a set of residues specified in the “uids” XML attribute of the “CorrectionBlock” XML element. Consider for example RESID:AA0185, 3-oxoalanine. This one modification can arise from either of two encoded amino acids, cysteine or serine. The first correction block, with uid=“AA0005”, gives the difference formula and weights based on the modification of cysteine,
(C 3 H 3 N 1 O 2 S 0) - (C 3 H 5 N 1 O 1 S 1) = (C 0 H -2 N 0 O 0 S -1)
and the second, with uid=“AA0016”, gives the difference formula and weights based on the modification of serine,
(C 3 H 3 N 1 O 2) - (C 3 H 5 N 1 O 2) = (C 0 H -2 N 0 O 0)
In this example, there are only losses from either starting residue, and the difference formulas and masses have negative values. Formulas always have positive values, but the correction or difference formulas can have positive or negative values.
|
Q: How are the Formula Block and Correction Block used?
A: These are primarily used for calculating the masses for mass spectrometry. The mass of a polypeptide can be calculated by adding the masses of all the residues in the sequence, then adding the weight of one water molecule (an H for the free alpha-amino end and an OH for the free alpha-carboxyl end), then adding the difference weights for modifications.
Alternatively, working backwards, you may be able to detect a modification by calculating the difference between the mass of the encoded peptide and the mass of the observed peptide, and looking for that mass difference among the mass differences in the RESID Database entries.
|
Q: Why are there “chemical” and “physical” weights? What is the difference?
A: The “chemical” and “physical” weight terms are a convenient short way of saying “the average isotope weight” and “the most common isotope weight”. The chemical weight is what would be observed by normal chemical methods, gel filtration or centrifugation, and the physical weight is what would be observed by mass spectrometry.
These terms hark back to the old days (in the 1950s) when there were two scales for atomic masses, a “chemical scale” used, oddly enough, by chemists and based on average elemental isotope abundances with the “average” oxygen assigned a mass of 16, and a “physical scale” used, you guessed it, by physicists and based on isotope masses with oxygen-16 assigned a mass of 16. Eventually they agreed to split the difference and define a single scale with carbon-12 assigned a mass of 12.
|
Q: How do I look in the RESID Database for all the known modifications with a particular mass difference?
A: Let us look for the modifications producing a mass difference of 80.00. Using the SRS search engine at http://www.ebi.ac.uk/resid/search.html or through SRS directly, in the selection box that shows “All fields” pull down and select “CWeightc” (“Correction weight chemical”). Then in the adjacent text entry box type “80”, and finally hit the “Search” button.
We get nothing; there are no modifications with a mass difference of precisely 80. We need to enter a range depending on how accurate our measurement of the mass difference is. If our measurement is 80.0 plus/minus 0.1, then in the text entry box type “79.9:80.1”.
We get all the modifications resulting either from replacement of an -H by -PO3H2, a mass difference of 79.98, or from replacement of an -H by -SO3H, a mass difference of 80.06 by an average isotope method.
If we have high-precision mass spectrometric measurements and our most common isotope mass difference is 79.9660 plus/minus 0.0005, then select “CWeightp” ("Correction weight physical"), and in the text entry box type “79.9655:79.9665”. We get all the modifications resulting from replacement of an -H by -PO3H2, a mass difference of 79.9663 by a most-common-isotope method.
|
Q: How are the entries in the RESID Database produced, and how frequently is the RESID Database updated?
A: The entries in the RESID Database are produced primarily from surveying literature reports of new protein modifications. Curators of the UniProt Knowledgebase at the Swiss Institute of Bioinformatics, at the European Bioinformatics Institute, and at the Protein Information Resource also encounter new modifications in the process of preparing feature annotations for proteins entries. Data-mining procedures are being used to look for modifications not yet documented in the RESID Database both from new literature reports as they appear and from older literature reports as they become available on-line.
Releases of the RESID Database have been produced at the end of every quarter since it was first publicly distributed in 1995. Interim updates are sometimes produced as frequently as once every two weeks, but usually two or three times in a quarter. The interim updates normally appear on Fridays, and the quarterly releases normally appear on the last day of the quarter. You may request to be sent E-mail notices for RESID Database updates by contacting
John Garavelli.
|
Q: How do I search the RESID Database for entries that were created or changed before or after a particular date?
A: Entries in the RESID Database have three date records, a creation date, a structure revision date, and a last text change date. New entries in the database have all three dates the same. The creation date never changes, but the structure revision and last text change dates are changed whenever the structure or text annotation of an entry is updated.
It is possible to search for entries based on these dates. The search engine uses the numeric date format yyyymmdd and either a single date or a range separated by a colon. To find the entries created during 2005, select the “CreationDate” field, and enter “20050101:20051231”.
Q: Where is the “controlled vocabulary” for the RESID Database?
A: The RESID Database of Protein Modifications itself documents in its
“Feature” records the standard annotations used to describe protein
modifications in the UniProt Knowledgebase feature records. Otherwise, the controlled vocabulary for the other record types in the RESID Database is included in the Document Type Definition file, RESID.DTD, which is available at the ftp sites.
Q: The formulas and masses do not seem to correspond to the amino acids. How are the formulas and masses calculated, and how should they be used?
A: The basic conceptual entities of the RESID Database are amino acid residues of polypeptides, that is the amino acid except for one proton from the alpha amino group and a hydroxyl from the alpha carboxyl group, equivalent to an amino acid less a water. There are two reasons for this. First, in order to calculate the molecular mass of an unmodified protein from its sequence and feature table, the best method is to sum the residue masses and add a proton and a hydroxyl (or 1 water) for each peptide chain. The other method sums the amino acid masses and subtracts N-1 waters. With the residue method, a water is already subtracted from each amino acid and one needs to be added back, unless the amino- or carboxyl terminals are modified. Second, the incremental differences in the b and y series in mass-spec correspond to the modified residue masses.
The correction masses for the modifications, the difference between the modified residue mass and the masses of the encoded residues must take into account three special conditions for each modification (listed in the "Condition" record) being amino-terminal (the proton correction is not required), being carboxyl-terminal (the hydroxyl correction is not required), or having a carboxamidine (an additional proton loss from a neighbouring residue in certain cross-links). The method for calculating modified protein masses from sequences and protein feature tables is then, sum the residue masses, add a proton and a hydroxyl (or 1 water) for each peptide chain, and add the correction masses for each modification (the correction masses take into account the fact that the added water will be too much if the amino- or carboxyl-terminal is modified).
For some modifications there are several ways in which the modification may arise, possibly from different encoded amino acids, and these will require different accounts and corrections. For example, cystine (RESID:AA0025) can arise either from a disulfide bond cross-linking two peptide chain cysteines, or from a peptide chain cysteine forming a disulfide bond with a free cysteine (which would not need the amino- and carboxyl-terminal accounting). For amino-terminal pyruvate (RESID:AA0127), there are also two correction cases, because this modification can arise from either serine or cysteine.
Q: If the process of methylation replaces a hydrogen with a methyl group giving a net change of (C 1 H 2) or +14.015650 Da, why is the difference formula for N,N-dimethylproline (C 2 H 5), and why are the difference formulas for N,N,N-trimethylalanine and N6,N6,N6-trimethyllysine given as (C 3 H 7), while the difference formula for Nω,Nω,Nω’-trimethylarginine is (C 3 H 6)?
A: When using the RESID Database to calculate the molecular weight of a peptide, we start with the neutral, uncharged residues and sum their formulas and masses. Next, the difference formulas and masses for the modifications are added. Unless there is an N-terminal modification, one hydrogen atom is added to complete the terminal amino group, and unless there is a C-terminal modification, one hydrogen and one oxygen are added to complete the terminal carboxyl group. Finally, any charges are introduced as required. In mass-spectrometry, it is usual to run in positive-ion analysis mode, so the neutral parent mass M is usually measured with one or more accompanying protons H+ and is written as MH+.
The uncharged amino group of the N-terminal or of an N6 lysine has only two hydrogen atoms for methyl replacement. When a third methyl group is added there is no third hydrogen to be replaced, and a methyl group must be added as the cation CH3+. When a quaternary amine is created, the intrinsic cation does not have an acidic proton that can transfer the charge. The difference formulas and masses are always measured from the same starting point, the neutral parent, so the difference formula must be (C 3 H 7) with a formal charge of 1+. If it is assumed that a measured ion is MH+, and a proton is subtracted to obtain a parent that is (C 3 H 6) more than the expected parent, the fact that the modified peptide may be intrinsically charged is being ignored, along with the chemical structure that is actually present. When the modified residue is intrinsically charged, the measured ion is M+, rather than MH+. The discrepancy comes down to how accounting is done for formal charges.
In the case of an N-terminal proline, only one hydrogen could be replaced in the neutral parent, and the second methyl must be a cation CH3+ creating a quaternary amine. In the case of trimethylarginine, three hydrogens are available in the parent for methyl replacement, and the modified residue is not an intrinsically charged quaternary amine.
[In PSI-MOD there are entries for both the difference from the neutral parent, and for the difference from the proton added, charged parent.]
|
|