 |
PIRSF Database
What is
PIRSF?
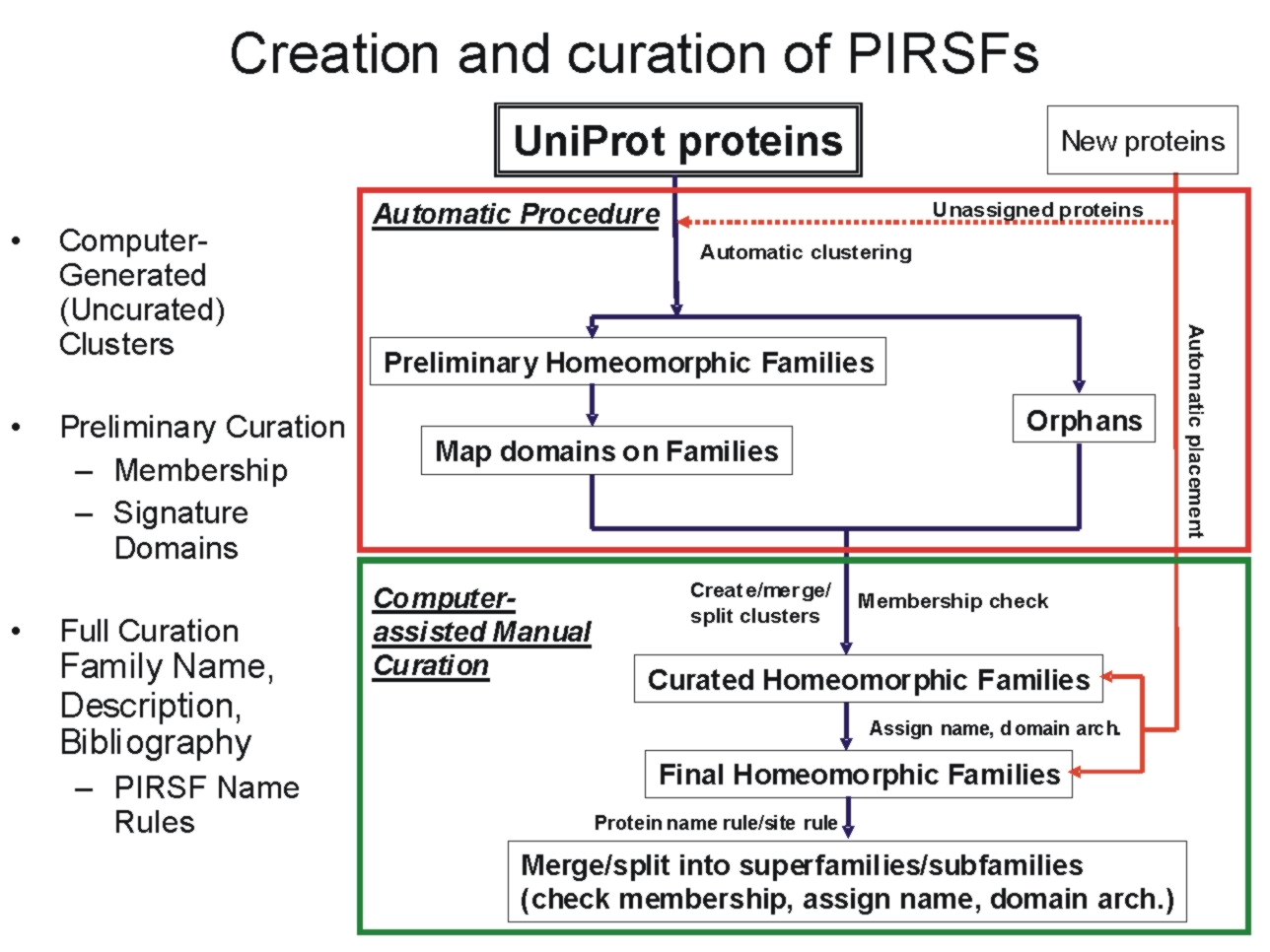
The PIRSF
protein classification
system is a network with multiple levels of
sequence diversity from superfamilies to
subfamilies that reflects the evolutionary
relationship of full-length proteins and domains.
The primary PIRSF classification unit is the
homeomorphic family, whose members are both
homologous (evolved from a common ancestor) and
homeomorphic (sharing full-length sequence
similarity and a common domain architecture).
Automatically generated protein clusters are
manually curated for membership, domain
architecture, annotation of sequence features, and
specific biological functions and biochemical
activities, when possible.
What in PIRSF for me?
PIRSF offers curated protein families with rules for functional site and protein name propagation and standardization, therefore, improving the sensitivity of protein identification and functional inference. Searching your protein sequence
against PIRSF database provides a faster and more accurate assessment of its function than a BLAST search against an
uncurated protein database. It avoids pitfalls such as numerous erroneous annotations, best hits based on a domain
secondary to the main protein function, spurious hits, etc.
PIRSF Definitions
-Curation Status
None: Computer-generated protein clusters, no manual curation. The clusters are computationally defined using both pairwise based parameters (% sequence identity, sequence length ratio and overlap length ratio) and cluster-based parameters (% matched members, distance to neighboring clusters and overall domain arrangement).
Preliminary: Computer-generated clusters are manually curated for membership (do proteins belong to the assigned cluster?) and domain architecture (Pfam domains listed from N- to C- termini).
Full/Full (with description): A name is assigned to the protein family, and accompanying references are listed when available. In many cases, brief descriptions are also provided.
-PIRSF Membership
Full (F): proteins
sharing end-to-end sequence similarity and common domain architecture.
Associate (A): members whose lengths are outside the family length range, including sequences
fragments, alternate splice and alternate initiator variants, and peptides derived from
proteolytic processing, are classified as associate members with the conceptual complete
sequence from which they are derived. Associate members also include individual proteins
with atypical domain architecture (thus, not yet forming a separate subfamily).
Seed (S): full members that are used to generate family specific
full-length and domain HMMs.
Use the following formats to perform a search: PIRSFxxxxxx:F, PIRSFxxxxxx:S or PIRSFxxxxxx:A for Full, Seed or Associate, respectively, with xxxxxx being the PIRSF number.
-PIRSF Name Evidence Tag
[Validated]: to indicate that at least one
member in the family has experimentally-validated function.
[Predicted]: for families whose functions
are inferred computationally based on sequence similarity and/or functional associative
analysis.
[Tentative]: cases where experimental evidence is not decisive.
-PIRSF Family Level
Homeomorphic family (HFam): members belonging to the family are both homologous (evolved from a common ancestor) and homeomorphic (sharing full-length sequence similarity and a common domain architecture). The homeomorphic family level is
the primary PIRSF curation level � and most significant in terms of annotation and most
invested with the biological meaning. A protein may be assigned to one and only one
homeomorphic family, which may have zero or more parent nodes and zero or more child
nodes.
Subfamily (SubFam): The subfamily level is used to delineate protein clusters within a
homeomorphic family that have specialized functions and/or variable domain
architectures. Like its parent, each subfamily is also homologous and homeomorphic. A
protein may be assigned to zero or one subfamily, which will have exactly one parent
node. The PIRSF IDs for subfamilies start with a number 5 (PIRSF5xxxxx).
Superfamily (SuperFam): The superfamily level is used to bring together a number of distantly
related families and orphan proteins that share one or more domains. Depending on the
extent of domain coverage, a superfamily may be a �homeomorphic superfamily�
(common domain architecture with full-length sequence coverage) or a �domain
superfamily� (partial sequence coverage). The PIRSF IDs for superfamilies start with a number 8 (PIRSF8xxxxx).
-Domain Architecture
It represents the curated information regarding present Pfam domains in the protein family. Pfam domains are listed in order from N- to C- termini separated by semi-colons. In a few cases, domains are separated by a dash indicating the presence of inserted domains.
Numbers in parenthesis indicate the repetition of a domain. There is a particular syntax for this feature. For example, PF11111(1-3) allows for 1 to 3 copies of PF11111, whereas PF11111(2-) allows for any number of domains above 1 (2 or more).However, PF11111(0,2) allows for none or two copies of this domain.
-Representative Sequence
It is the sequence of a member of a PIRSF that belongs to a model organism (if present), belongs to UniProtKB/Swiss-Prot (if any) and meets the PIRSF criteria of uniform length and common domain architecture. Representative sequences are assigned automatically, but curator may decide to change it.
iProClass Database
What is iProClass?
iProClass
provides summary descriptions of protein family,
function and structure for UniProt
sequences, with links to over 90 biological
databases. iProClass comprises reports for all UniProtKB proteins and those proteins that are exclusively in UniParc database.
iProClass
Text Search
Retrieve a matching list of
summary reports by text string or unique
identifier (selecting a field from the
dropdown menu). Click "Search" button to retrieve
results. You may open extra input boxes in your
query by clicking on "Add input box". See Text
Search Help for additional
information.
iProLINK Help
What is iProLINK?
As PIR focuses its effort on the curation of the UniProtKB protein sequence database, the goal of iProLINK is to provide curated data sources that can be utilized for text mining research in the areas of bibliography mapping, annotation extraction, protein named entity recognition, and protein ontology development.
The process of applying literature mining methods for protein database curation involves several tasks:
- Bibliography Mapping: identification of articles from literature sources (such as PubMed) that describe a given protein entry;
- Annotation Extraction: categorization of annotation types and extraction of sentences and/or phrases describing the given annotation; and
- Database Curation: conversion of the extracted literature information into annotation in the database with structured syntax, controlled vocabulary, and evidence attribution.
|
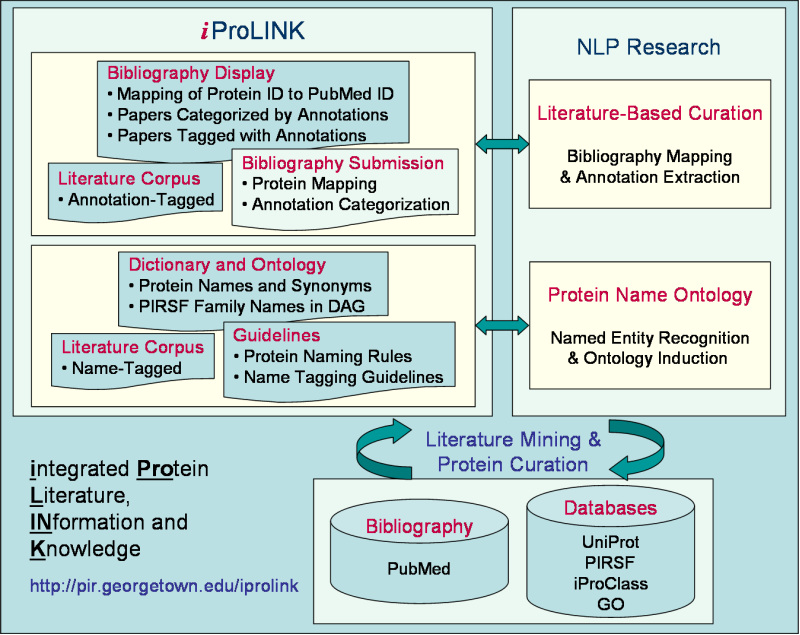 |
These tasks are also related to the topics of protein named entity recognition and protein ontology development.
A prerequisite to bibliography mapping is protein named entity recognition/identification of protein names from articles.
Furthermore, due to the long-standing problem of protein nomenclature, a protein ontology can assist entity recognition with the description of names and synonyms of protein classes as well as their relationships.
Bibliography Mapping
Linking protein entries to relevant scientific literature that describes or characterizes the proteins is crucial for increasing
the amount of experimentally verified data and for improving the quality of protein annotation.
Enter a text string or a unique identifier to retrieve bibliography related to you query.
Results are shown in a table similar to iProClass result page, but with specific columns related to bibliography retrieval. Links to the bibliography record as well as the individual PubMed entries are available.
Feature Evidence Attribution
In the PIR-PSD database, feature annotation such as binding sites, catalytic sites, and modified sites are labeled with status tags "experimental" or "predicted" to distinguish experimentally verified from computationally predicted data. To appropriately attribute bibliographic data to features with experimental evidence,
a retrospective literature survey was conducted, which involves both citation mapping (finding citations from the Reference section that describe the given experimental feature) and evidence tagging (tagging the sentences providing experimental evidence in an abstract and/or full-text article). Now this is being extended to UniProtKB proteins.
Search for proteins with a particular feature attribution such as post- translational modifications, or enter a UniProtKB identifier to retrieve feature attribution for your query.
Later on you may get curated bibliography related to the entry by clicking on "Bibliography" or all available tagged evidence by clicking on "Tagged Evidence"
Examples for Feature Evidence Attribution Text Search
| Feature |
Example |
| Active site | ser |
| Binding site | heme |
| Cleavage site | arg |
| Cross-link | cys |
| Disulfide bonds | all |
| Modified site | block* |
| Domain | signal sequence |
| Product | dermorphin |
| Region | ATP binding |
Entity Recognition/Ontology Development
Protein named entity recognition (finding protein names from literature texts) is a prerequisite for bibliography mapping (identifying papers describing specified proteins). It is also fundamental for several other biological literature mining tasks, including the extraction of protein annotations (such as protein-protein interactions) from literature.
-Protein Name Dictionary and Word Token Dictionaries
PIR protein name dictionary is derived from the protein name field in the iProClass database, which consists of protein names from UniProt (Swiss-Prot,TrEMBL, PIR-PSD) and RefSeq. After the initial compilation, the dictionary undergoes several filtering processes to generate unique protein names (including synonyms and acronyms), and to remove nonsensical names and certain general descriptional annotations. For example, entry names such as "Inter-alpha-trypsin inhibitor (GIK-14) (Fragment)" were broken into Inter-alpha-trypsin inhibitor, GIK-14 and Fragment. The name Fragment is later removed from the dictionary along with a list of other "bad" names such as hypothetical protein, conserved hypothetical protein, unnamed protein product, predicted protein, and predicted protein of unknown function. In addition, words such as probable, putative, and similar to before protein names are also removed so that a name like putative aspartate aminotransferase A is merged to aspartate aminotransferase A to reduce the redundancy. Derived from over 1.5 million iProClass entries, the protein name dictionary currently has about 700,000 names, each of which is shown with its frequency count.
Most protein names are composed of combinations of two or more words (or tokens). Therefore, protein name rules can be derived from tokenized protein words and used during post-tagging processing to improve machine learning-based named entity recognition. We have compiled specialized single-word dictionaries by tokenization and classification of protein names from 30,000 well-curated iProClass protein entries (each containing at least 5 reference citations). The dictionaries consist of individual word tokens categorized into five classes.
- Biomedical Terms (bt): These terms are used in a broad of biological and medical sciences. They mainly describe structures of all forms of life at different levels (from gross morphology to molecular structure), as well as their respective functions and mechanisms in both normal (physiological) and diseased states (pathological).
- Chemical Terms (ct): These are words that describe organic or inorganic chemical materials, chemical groups or bonds, or chemical properties.
- Macromolecules (mc): These words refer to biopolymers such as proteins, peptides, DNA, RNA, polysaccharides, or glycoproteins.
- Common English (ce): Common English words are used to describe various aspects or properties of proteins, such as short, signal, interacting, and repair. These also include spelled-out forms of Greek letters, as well as stop words like of, at, and to.
- Non-word tokens: They are combinations of letters, numbers, or symbols. They often are acronyms, synonyms, or abbreviations. The form of non-word tokens can be number only, single letter, multiple letters, or combinations of numbers, letters, and other symbols. Non-word tokens may stand for biochemical entities such as nucleic acids, nucleotide, and amino acids, which can be used in a protein name.
-Protein Name Tagging Guidelines and Name-Tagged Corpora
Other iProLINK data resources for named entity recognition are two sets of literature corpora that were manually tagged with protein names based on two versions of tagging guidelines. Guideline 1.0 defines how to tag protein objects, not protein named entities, whereas Guideline 2.0 defines tagging rules for protein named entities regardless of the context of the object.
-PIRSF Family Classification-Based Protein Ontology
Biological ontologies are crucial for biological knowledge management, including mining literature data to extract relevant information and integrating information from multiple databases. A protein ontology?consisting of names and synonyms of protein classes as well as their relationships?can be used to assist with protein named entity recognition. Furthermore, an ontology based on protein family relationships, such as the PIRSF classification system, can be mapped to and complement the Gene Ontology (GO).
We have developed a protein ontology based on PIRSF hierarchical family names. The ontology is in the GO flat file format with a DAG (directed acyclic graph) structure, and can be browsed using application tools such as DAG-Edit.
Text Search Help
Select a
Database
Depending on the route by which
the Text Search page was accessed, you may need to
select between the iProClass and PIRSF databases. In the former case your query will be searched against UniProtKB and unique UniParc proteins. In the latter case, it will be searched against the whole set of PIRSF
families (i.e., any curation level). In the home page, Text Search is performed using the iProClass database.
Selecting a
Field
Searchable
fields can be selected from a dropdown menu.
The items will vary according to database
selected, since each database contains different
types of information.
Query Input
Enter a unique
identifier or other search string in the box
provided.
Certain items (such as "Length" or
anything with "ID") will be exact match searches.
Other items will be substring searches (as if
preceded and followed by wild cards).
When the
field option is present, entering "not null" in
the text box will cause the search to return only
those entries that have some data in the selected
field, while entering "null" will return only
those that lack data in the selected field.
For
peptide search, type in a string of amino acid
residues in single
letter code (at least three letters), then
press the arrow to retrieve results.
Add Input Box Button
If
desired, multiple fields can be searched
simultaneously. Pressing the "+ box" button adds
another query line, up to a maximum of 8 lines.
The added input boxes are connected by logical
operator choices (see below), with the default
being the Add operator.
Using the Logical Operators AND,
OR, NOT
Search supports the logical 'AND',
'OR', and 'NOT' operators. For example, to
retrieve results that include Pfam domains A or B,
type A in your first query field and add a query
line by clicking the "Add input box" button. Enter
your 2nd query (B) and select the OR operator.
Similarly, to retrieve multi-domain proteins that
have both Pfam domains A and B, use the 'AND'
operator. Proteins that have domain A and not
domain B can be retrieved using the 'NOT'
operator.
Number of
Results
To provide the fastest
result, the default number of entries shown
on any one page is 50.
Search
Categorizations and Unique Identifiers
The
following table indicates the searchable fields
for Text and Batch Retrieval search
functions. Searchable fields can be selected from
the dropdown menu. You can search the databases
using unique identifiers or keywords. Entry
examples are shown below.
Text Search Result Help
This section describes the Text Search Result page for iProClass and PIRSF. The overall layout of the page is similar for both, however, it differs in the default columns that are displayed, as well as the tools available to analyze data.
Common features are described first 1-3, where numbers indicate the place where you will find these features in the page (see iProClass and PIRSF result pages below).
1- Search
This feature allows you to perform an additional search in case you want to further filter out your output
or you want to start a new search (no need to go back to the previous page unless you want to use a different database).
2- Display Options
Depending on your specific need you can
choose the columns to be displayed. To do this, click on the "Display Option" button, select the
relevant field(s) in the "Fields Not in Display"
list and transfer them to the "Fields in
Display" list via the ">" button. Conversely,
columns can be removed from display. Finally, click on "apply"
for the changes to take effect.
3- Save Results
As
The output can be saved to the
user's local computer. The results will be saved
for selected entries or, if no proteins are
selected, for all entries. Clicking "Table" will
save the displayed columns as a tab-delimited text
file, which may be imported into a spreadsheet for
easier viewing or analysis. Clicking "FASTA" will
save the IDs and sequences in FASTA
format.
iProClass Result Page
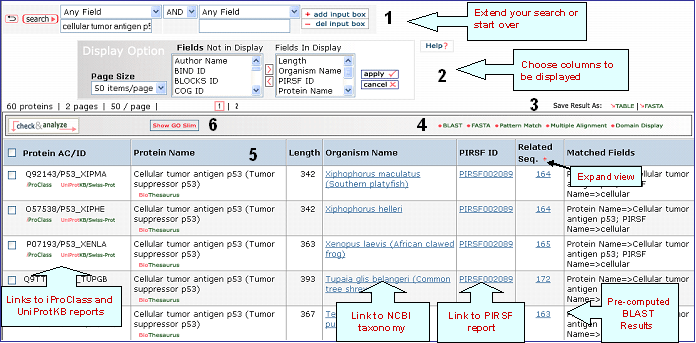
4- Analyze: BLAST,
FASTA, Pattern Match, Multiple Alignment and
Domain Display
Retrieved entries can be
further analyzed using the sequence analysis
programs available in the Results page. First,
select the protein(s) using the checkboxes on the
left side of the table, then click the
corresponding analysis tool. Click "BLAST" or
"FASTA" button, and a new query page will be
displayed, along with the parameters that were
selected in the initial search. Click "Pattern
Match" to search against the PROSITE database. For
multiple alignment, check at least 2 proteins (but
no more than 50), then click the "Multiple
Alignment" button. A ClustalW alignment
and neighbor-joining tree will be generated along with a link to the interactive PIR viewer.
Domain display option, shows PFam domains (if
present) in graphical format.
5- Results Display
Results of
the search are displayed in a customizable table.
The exact columns displayed will depend on the fields searched for, and
user preference.
Protein AC/ID
The
Protein AC/ID refers to the UniProtKB or UniParc identifiers. Below these numbers, you may choose either the iProClass or the UniProtKB/UniParc view of the protein report. The source of the UniProtKB sequence is shown as UniProtKB/Swiss-Prot or UniProtKB/TrEMBL if the protein sequence is from Swiss-Prot or TrEMBL section, respectively. Alternatively, the UniParc ID will be displayed if the sequence is no present in the UniProtKB database along with the UniParc report.
Protein Name
The common
name given to a protein, that identifies its
function or specifies its features.
Length
Number of amino acid
residues in the peptide or protein.
Organism Name
The genus and species of the source organism from which the sequence originated. Links to NCBI taxonomy information are provided.
PIRSF ID
If a protein belongs to a PIRSF family, then this column will display the corresponding family identifier. Click on the ID to retrieve the PIRSF report (see annotated output).
Related Seq.
This column shows the number of pre-computed BLAST hits obtained using default parameters. Only up to 300 sequences will be displayed. By clicking the number you access to the related sequence page. This allows to have a glance at sequence similarity in a very fast way. The number itself already provides some information about how unique the protein is. For example, a very low number may tell you that the query is specific to a certain species, genus, taxon, etc. The "+" sign next to the Related Sequence title allows to compare number of related sequences at 3 different E-value cut-offs as shown below.

Matched
Fields
The field(s) matched by the
query.
PIRSF Result Page
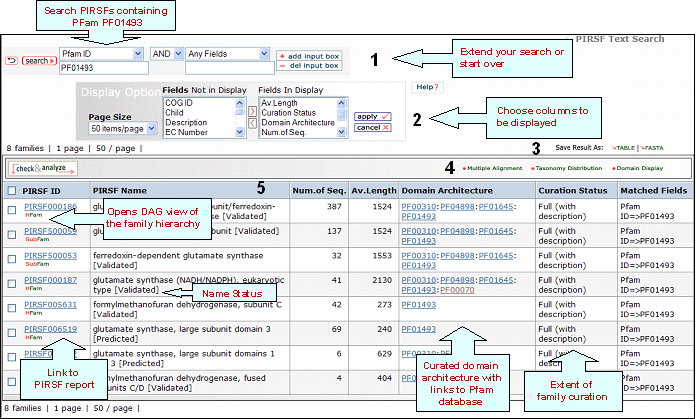
4- Analyze: Multiple Alignment, Taxonomic Distribution and
Domain Display
Retrieved entries can be
further analyzed using the sequence analysis
programs available in the Results page. First,
select the PIRSF(s) using the checkboxes on the
left side of the table, then click the
corresponding analysis tool. Click on the "Multiple
Alignment" button to generate a ClustalW alignment
and neighbor-joining tree for the members of the selected PIRSF. If one PIRSF is selected, then alignment of all its seed members will be performed (see sample output for PIRSF000186). But if many PIRSFs are selected, then alignment of the representative sequences will be displayed. Click on Taxonomic Distribution to look at the number of members of a PIRSF present in the different taxa. You can perform this function on one or multiple PIRSFs. If only one PIRSF is selected, the taxonomic distribution for the parent family and the children (if any) will be displayed. See sample output for PIRSF000186.
Domain display option, shows PFam domains (if
present) in a graphical format.
5- Results Display
The results of
the search are displayed in a customizable table.
The exact columns displayed will depend on the fields searched for, and
user preference.
PIRSF ID
This column will display the corresponding family identifier. Click on the ID to retrieve the PIRSF report. The icon below the PIRSF ID states the family level, namely homeomorphic family (HFam), subfamily (SubFam), and superfamily (SuperFam). Clicking on this icon will bring the DAG view of PIRSF Hierarchy with the PFam domain at the higher level.
PIRSF Name
The
name given to the family that identifies its
function or specifies its features. Depending on the curation status, this name is assigned automatically (no manual curation) or manually (full curation). If the family is curated, it may display a tag next to the name indicating its status, namely validated (there is experimental data supporting PIRSF name), tentative (there is no conclusive evidence) or predicted (predicted by computational methods).
Av.Length
It is the protein average length of the members of the PIRSF.
Domain Architecture
It represents the curated information regarding present Pfam domains in the protein family. Pfam domains are listed in order from N- to C- terminus separated by semi-colons. In a few cases, domains are separated by a dash indicating the presence of inserted domains.
Numbers in parenthesis indicate the repetition of a domain. There is a particular syntax for this feature. For example, PF11111(1-3) allows for 1 to 3 copies of PF11111, whereas PF11111(2-) allows for any number of domains above 1 (2 or more). However, PF11111(0,2) allows for none or two copies of this domain.
Curation Status
None: Computer-generated
protein clusters, not
curated.
Preliminary: Membership and
domain architecture of protein families determined
by manual curation.
Full:Protein family
name with accompanying references (when
available), and sometimes brief descriptions,
provided after thorough manual curation.
Matched
Fields
The field(s) matched by the
query.
Batch Retrieval
Help
Retrieve multiple entries
by selecting a specific identifier or a combination of identifiers.
Select the database
Before entering the protein identifiers you need to select the most convenient database. If you want to retrieve information about protein families, then select the PIRSF database, however if you are interested in analyzing individual proteins, then select the iProClass database.
Rules for entering
IDs
Multiple Entry
IDs should be separated by lines or
spaces.
The maximum numbers of entries allowed
is 50.
IDs may be specified as a single
category or as mixed categories. However, if your entries have the same type of ID, it is recommended that you define the ID field to speed up the retrieval process.
You can further analyze and save any
retrieved results and/or their respective
sequences in a similar way as you do after a Text Search. Click on "Show Match List" to check the correspondence between your entry identifiers and the ones from the selected database.
Batch Retrieval Output in iProClass Database
Retrieval of sequences with GI numbers 1169968, 1707983 and 304131
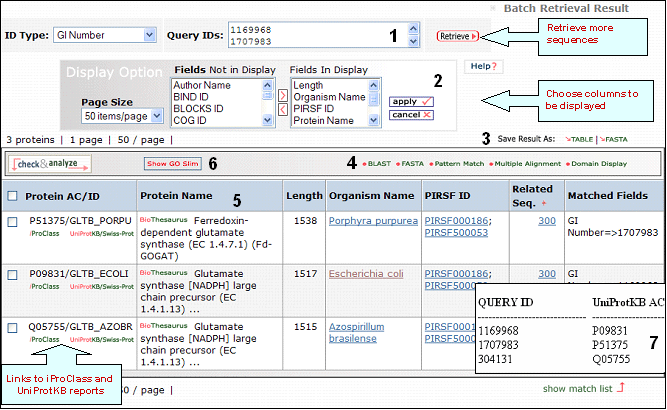
1- Retrieval Box
This box shows your query ID and also allows you to perform a new retrieval.
2- Display Options
Depending on your specific need you can
choose the columns to be displayed. To do this, click on the "Display Option" button, select the
relevant field(s) in the "Fields Not in Display"
list and transfer them to the "Fields in
Display" list via the ">" button. Conversely,
columns can be removed from display. Finally, click on "apply"
for the changes to take effect.
3- Save Results
As
The output can be saved to the
user's local computer. The results will be saved
for selected entries or, if no proteins are
selected, for all entries. Clicking "Table" will
save the displayed columns as a tab-delimited text
file, which may be imported into a spreadsheet for
easier viewing or analysis. Clicking "FASTA" will
save the IDs and sequences in FASTA
format.
4- Analyze: BLAST,
FASTA, Pattern Match, Multiple Alignment and
Domain Display
Retrieved entries can be
further analyzed using the sequence analysis
programs available in the Results page. First,
select the protein(s) using the checkboxes on the
left side of the table, then click the
corresponding analysis tool. Click "BLAST" or
"FASTA" button, and a new query page will be
displayed, along with the parameters that were
selected in the initial search. Click "Pattern
Match" to search against the PROSITE database or against a user defined pattern. For
multiple alignment, check at least 2 proteins (but
no more than 50), then click the "Multiple
Alignment" button. A ClustalW alignment
and neighbor-joining tree will be generated along with a link to the interactive PIR viewer.
Domain display option, shows PFam domains (if
present) in graphical format.
4- Results Display
Results of
the search are displayed in a table.
Protein AC/ID
The
Protein AC/ID refers to the UniProtKB accession number and id, respectively. Below these, you may choose either the iProClass or the UniProtKB/UniParc view of each protein report. The source of the UniProtKB sequence is shown as UniProtKB/SP or UniProtKB/Tr if the protein sequence is from Swiss-Prot or TrEMBL, respectively.
Protein Name
The common
name given to a protein, that identifies its
function or specifies its features.
Length
Number of amino acid
residues in the peptide or protein.
Organism Name
The genus and species of the source organism from which the sequence originated. Links to NCBI taxonomy information are provided.
PIRSF ID
If a protein belongs to a PIRSF family, then this column will display the corresponding family identifier. Click on the ID to retrieve the PIRSF report.
Match
Range
This column displays in red the peptide query within the sequence.
6- Show match list
Shows a table mapping your query IDs with the UniProtKB/UniParc IDs.
Batch Retrieval Output in PIRSF Database
Retrieval of PIRSFs using as queries UniProtKB ACs P51375, P09831 and Q05755
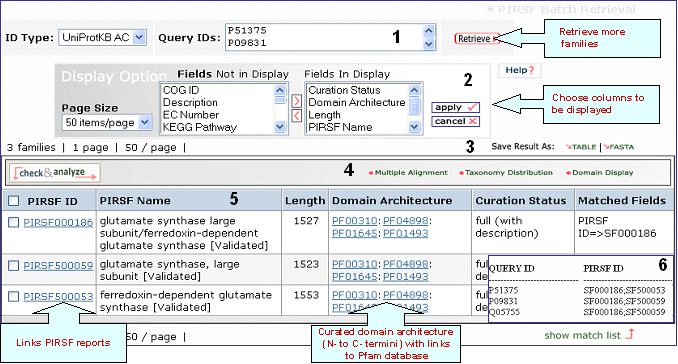
1- Retrieval Box
This box shows your query ID and also allows you to perform a new retrieval
2- Display Options
Depending on your specific need you can
choose the columns to be displayed. To do this, click on the "Display Option" button, select the
relevant field(s) in the "Fields Not in Display"
list and transfer them to the "Fields in
Display" list via the ">" button. Conversely,
columns can be removed from display. Finally, click on "apply"
for the changes to take effect.
3- Save Results
As
The output can be saved to the
user's local computer. The results will be saved
for selected entries or, if no proteins are
selected, for all entries. Clicking "Table" will
save the displayed columns as a tab-delimited text
file, which may be imported into a spreadsheet for
easier viewing or analysis. Clicking "FASTA" will
save the IDs and sequences in FASTA
format.
4- Analyze: Multiple Alignment, Taxonomic Distribution and
Domain Display
Retrieved entries can be
further analyzed using the sequence analysis
programs available in the Results page. First,
select the PIRSF(s) using the checkboxes on the
left side of the table, then click the
corresponding analysis tool. Click on the "Multiple
Alignment" button to generate a ClustalW alignment
and neighbor-joining tree for the members of the selected PIRSFs. If one PIRSF is selected, then alignment of all its seed members will be performed (see sample output for PIRSF000186). But if many PIRSFs are selected, then alignment of the representative sequences will be displayed. Click on Taxonomic Distribution to look at the number of members of a PIRSF present in the different taxa. You can perform this function on one or multiple PIRSFs. If only one PIRSF is selected, the taxonomic distribution for the parent family and the children (if any) will be displayed. See sample output for PIRSF000186.
Domain display option, shows PFam domains (if
present) in a graphical format. See sample output or annotated output for PIRSF000186.
5- Results Display
Results of
the search are displayed in a table.
PIRSF ID
This column will display the corresponding family identifier. Click on the ID to retrieve the PIRSF report. The icon below the PIRSF ID states the family level, namely homeomorphic family (HFam), subfamily (SubFam), and superfamily (SuperFam). Clicking on this icon will bring the DAG view of PIRSF Hierarchy with the PFam domain at the higher level.
PIRSF Name
The
name given to the family that identifies its
function or specifies its features. Depending on the curation status, this name is assigned automatically (no manual curation) or manually (full curation). If the family is curated, it may display a tag next to the name indicating its status, namely validated (there is experimental data supporting PIRSF name), tentative (there is no conclusive evidence) or predicted (predicted by computational methods).
Length
It is the protein average length of the PIRSF members.
Domain Architecture
It represents the curated information regarding present Pfam domains in the protein family. Pfam domains are listed in order from N- to C- terminus separated by semi-colons. In a few cases, domains are separated by a dash indicating the presence of inserted domains.
Numbers in parenthesis indicate the repetition of a domain. There is a particular syntax for this feature. For example, PF11111(1-3) allows for 1 to 3 copies of PF11111, whereas PF11111(2-) allows for any number of domains above 1 (2 or more). However, PF11111(0,2) allows for none or two copies of this domain.
Curation Status
None: Computer-generated
protein clusters, not
curated.
Preliminary: Membership and
domain architecture of protein families determined
by manual curation.
Full:Protein family
name with accompanying references (when
available), and sometimes brief descriptions,
provided after thorough manual curation.
Matched
Fields
The field(s) matched by the
query.
6- Show match list
Shows a table mapping your query IDs to the corresponding PIRSF.
BLAST Search Help
What is BLAST?
The Basic Local
Alignment Search Tool (BLAST) allows rapid
sequence comparison that optimizes the
high-scoring segment pair (HSP), a measure of
local similarity. For more information visit NCBI-BLAST.
Select a Database
BLAST search can be performed in two databases: UniProtKB is the central hub for the collection of functional information on proteins, with accurate, consistent, and rich annotation. It consists of two sections: a section containing manually-annotated records with information extracted from literature and curator-evaluated computational analysis (UniProtKB/Swiss-Prot), and a section with computationally analyzed records that await full manual annotation (UniProtKB/TrEMBL).
UniRef100 provides clustered sets of sequences at 100 % from UniProt Knowledgebase (including splice variants and isoforms) and selected UniParc records (mainly from PDB, Refseq and Ensembl databases), in order to obtain complete coverage of sequence space while hiding redundant sequences (but not their descriptions) from view. It combines identical sequences and sub-fragments with 11 or more residues (from any organism) into a single UniRef entry, displaying the sequence of a representative protein, the accession numbers of all the merged UniProtKB entries, and links to the corresponding UniProtKB and UniParc records.
.
BLAST Search Format
The
BLAST 'Search' input area accepts:
">" followed by a UniProtKB sequence identifier. Spaces between letters in
the input are not allowed, although spaces before
or after the identifier are allowed.
Raw amino
acid sequence format:MSEPQRLFFAIDLPAEIREQIIHWRATHFPPEAGRPVAADNLHLTLAFLGEVS
AEKEKALSLLAGRIRQPGFTLTLDDAGQWLRSRVVWLGMRQPPRGLIQLAN
MLRSQAARSGCFQSNRPFHPHITLLRDASEAVTIPPPGFNWSYAVTEFTLYA
SSFARGRTRYTPLKRWALTQ
The query
sequence intersperse with numbers and/or spaces, such as:
1 msepqrlffa idlpaeireq iihwrathfp peagrpvaad nlhltlaflg evsaekekal
61 sllagrirqp gftltlddag qwlrsrvvwl gmrqpprgli qlanmlrsqa arsgcfqsnr
121 pfhphitllr daseavtipp pgfnwsyavt eftlyassfa rgrtrytplk rwaltq
Sequences are
expected to be represented in the standard
IUB/IUPAC amino acid codes, with these exceptions:
lower-case letters are accepted and are mapped
into upper-case; U and * are acceptable letters
(see table).
Numerical digits in the query sequence are
automatically removed.
BLAST
Options
BLAST is performed using the BLOSUM62 matrix with default values for gap opening and extension cost. However, the
following parameters can be set:
-Composition-based Statistics
BLAST permits calculated E-values to take into account the amino acid composition of the individual database sequences involved in reported alignments. This improves E-value accuracy, thereby reducing the number of false positive results. The improved statistics are achieved with a scaling procedure that in effect employs a slightly different scoring system for each database sequence. As a result, raw BLAST alignment scores in general will not correspond precisely to those implied by any standard substitution matrix. Furthermore, identical alignments can receive different scores, based upon the compositions of the sequences they involve.
-Filter
Low-complexity
Masks off segments of the query sequence that have low compositional complexity, as determined by the SEG program of Wootton & Federhen (Computers and Chemistry, 1993). Filtering can eliminate statistically significant but biologically uninteresting reports from the BLAST output (e.g., hits against common acidic-, basic- or proline-rich regions), leaving the more biologically interesting regions of the query sequence available for specific matching against database sequences. Filtering is only applied to the query sequence, not to database sequences.
Mask for Lookup Table Only
This option masks only for purposes of constructing the lookup table used by BLAST. BLAST searches consist of two phases, finding hits based upon a lookup table and then extending them. The option to "Mask for lookup table only" masks only for the lookup table so that no hits are found based upon low-complexity sequence. The BLAST extensions are performed without masking and so they can be extended through low-complexity sequence. This option is still experimental and may change in the near future.
Mask Lower Case
With this option selected you can cut and paste a FASTA sequence in upper case characters and denote areas you would like filtered with lower case. This allows you to customize what is filtered from the sequence during the comparison to the BLAST databases.
-Expect
The Expect threshold establishes a statistical significance threshold for reporting database sequence matches. The default value is 10, meaning that 10 matches are expected to be found merely by chance. Lower Expect thresholds are more stringent, leading to fewer chance matches being reported. Increasing the expected threshold shows less stringent matches and is recommended when performing searches with short sequences as a short query is more likely to occur by chance in the database than a longer one, so even a perfect match (no gaps) can have low statistical significance and may not be reported. Increasing the Expect threshold allows you to look farther down in the hit list and see matches that would normally be discarded because of low statistical significance.
-Word Size
The word size indicates the length of the initial sequence that must be matched between the database and the query sequence.
-Matrix
A key element in evaluating the quality of a pairwise sequence alignment is the "substitution matrix", which assigns a score for aligning any possible pair of residues. The matrix used in a BLAST search can be changed depending on the type of sequences you are searching with. The user may choose from a list of matrices that cover various evolutionary constraints (more information can be found in a description of BLAST scoring matrices). For each matrix, a default matrix-dependent gap cost is displayed. Gap costs are described below.
-Matrix-dependent Gap Cost
The pull down menu shows the Gap Costs (penalty to open gap and penalty to extend gap). There are a limited number of options for these parameters. Increasing the Gap Costs will result in alignments that decrease the number of Gaps introduced. The gap open penalty is the score taken away for the initiation of a gap in a sequence. To make the match more significant the user can try making the gap penalty larger. The gap extension penalty is added to the gap open penalty for each residue in the gap, effectively penalizing longer gaps. If the user does not like long gaps, they can increase the extension gap penalty. Usually one would expect a few long gaps rather than many short gaps, so the gap extension penalty should be lower than the gap penalty. An exception is where one or both sequences are single reads with possible sequencing errors, in which case you would expect many single base gaps. The user can get this result by setting the gap open penalty to zero (or very low) and using the gap extension penalty to control gap scoring.
-Adjust Gap Costs
Alignments between sequences are often optimized by allowing gaps within one or both sequences. Like mismatches between aligned residues, gaps have a "cost" associated with them. There are separate penalties to open and to extend gaps. Increasing the Gap Costs will result in alignments that decrease the number and size of Gaps introduced. The Gap Open cost (or Gap Existence cost) is the score taken away for the initiation of a gap in a sequence. To make the match more significant the user can try making this gap penalty larger. The Gap Extend cost is added to the Gap Open cost for each residue in the gap, effectively penalizing longer gaps. The user can therefore select against long gaps by increasing this penalty. Usually one would expect a few long gaps rather than many short gaps, so the Gap Extend cost should be lower than the Gap Open cost. The Gap Costs can be adjusted relative to the default value using the pull down menu.
-Number of Hits to Display
Restricts the number of BLAST hits of matching sequences that will be reported.
-Alignment
Aligns your query sequence and database matches in pairs. Matches are connected with a "|" symbol. Mismatches are opposed with a space. Gaps are introduced with a "-" symbol.
References
Wootton JC, and Federhen S (1993) Statistics of local complexity in amino acid sequences and sequence databases. Computers and Chemistry 17:149-163.
BLAST Results Help
A sample output for a BLAST search against the UniProtKB database is shown below
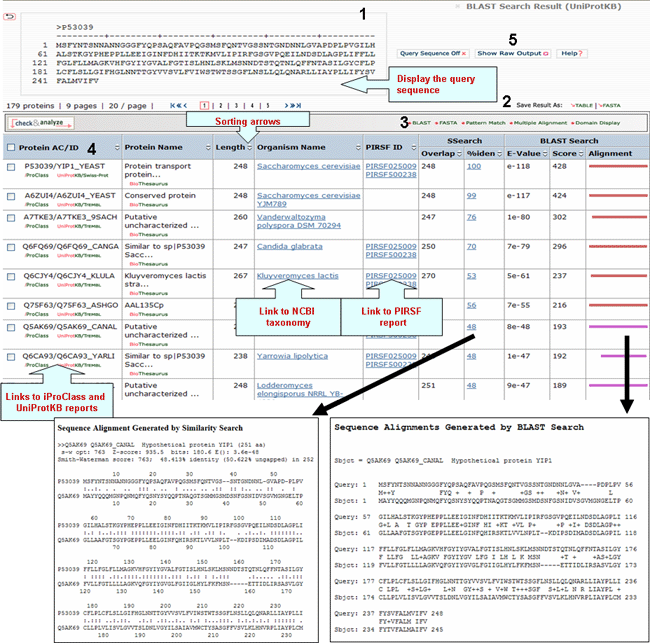
1- Query Sequence on
Click on this button to display the query sequence. Conversely, click it again to hide it.
2- Save Results As
Search results can be saved to the user's local computer. The results will be saved for selected entries or, if no proteins are selected, for all entries. Clicking "Table" will save the displayed columns as a tab-delimited text file, which may be imported into a spreadsheet for easier viewing or analysis. Clicking "FASTA" will save the IDs and sequences in FASTA format.
3- BLAST,
FASTA, Pattern Match, Multiple Alignment and
Domain Display
Retrieved entries can be
further analyzed using the sequence analysis
programs available in the Results page. First,
select the protein(s) using the checkboxes on the
left side of the table, then click the
corresponding analysis tool. Click "BLAST" or
"FASTA" button, and a new query page will be
displayed, along with the parameters that were
selected in the initial search. Click "Pattern
Match" to search against the PROSITE database. For
multiple alignment, check at least 2 proteins (but
no more than 50), then click the "Multiple
Alignment" button. A ClustalW alignment
and neighbor-joining tree will be generated along with a link to the interactive PIR viewer.
Domain display option, shows PFam domains (if
present) in graphical format.
4- Results
Display
Sort Columns
Columns can be
sorted by the corresponding values by clicking on
the arrow next to the column title. By default
the table is sorted by Score.
The results from the search are displayed
in a table with the following default columns:
Protein AC/ID
The
Protein AC/ID refers to the UniProtKB accession number and ID, respectively. Below these identifiers, you may choose either the iProClass or the UniProtKB view of the protein report. The source of the UniProtKB sequence is shown as UniProtKB/Swiss-Prot or UniProtKB/TrEMBL if the protein sequence is from the Swiss-Prot or TrEMBL section of UniProtKB, respectively.
Protein
Name
The common name given to a protein,
that identifies its function or specifies its
features.
Length
Number of amino acid
residues in the peptide or protein.
Organism Name
The genus and species of the source organism from which the sequence originated. Links to NCBI taxonomy information are provided.
PIRSF ID
If a protein belongs to a PIRSF family, then this column will display the corresponding family identifier. Click on the ID to retrieve the PIRSF report (see annotated output).
BLAST
Sequence Similarity Columns
There are three
columns related to BLAST results. The
Alignment column, in the extreme right, shows the
BLAST results in graphical format. The top
bar represents the query sequence. The bars below
show the region(s) on other sequences matched to
the query sequence. The bar color indicates the
magnitude of the BLAST score. Click on one of
these bars to see its BLASTalignment paired
with the query sequence.
SSearch Columns
SSearch is
a pairwise implementation of the Smith-Waterman
alignment algorithm. When two sequences are
aligned, only the shared region is shown. Within
the shared region, amino acid residues from one or
both sequences can be aligned with either amino
acids or gaps from the other sequence. The total
length of the shared region, including gaps, is
represented under the Overlap column. The
percentage of identical residues in the alignment
is given under %iden. Clicking on that number
displays the SSearch full-length
alignment.
A sample output for a BLAST search against the UniRef100 database is shown below
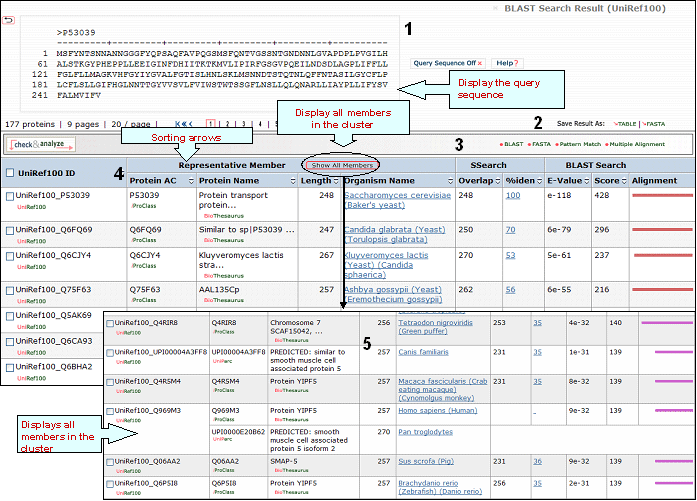
1- Query Sequence on
Click on this button to display the query sequence. Conversely, click it again to hide it.
2- Save Results As
Search results can be saved to the user's local computer. The results will be saved for selected entries or, if no proteins are selected, for all entries. Clicking "Table" will save the displayed columns as a tab-delimited text file, which may be imported into a spreadsheet for easier viewing or analysis. Clicking "FASTA" will save the IDs and sequences in FASTA format.
3- BLAST,
FASTA, Pattern Match, Multiple Alignment and
Domain Display
Retrieved entries can be
further analyzed using the sequence analysis
programs available in the Results page. First,
select the protein(s) using the checkboxes on the
left side of the table, then click the
corresponding analysis tool. Click "BLAST" or
"FASTA" button, and a new query page will be
displayed, along with the parameters that were
selected in the initial search. Click "Pattern
Match" to search against the PROSITE database. For
multiple alignment, check at least 2 proteins (but
no more than 50), then click the "Multiple
Alignment" button. A ClustalW alignment
and neighbor-joining tree will be generated along with a link to the interactive PIR viewer.
Domain display option, shows PFam domains (if
present) in graphical format.
4- Results
Display
Sort Columns
Columns can be
sorted by the corresponding values by clicking on
the arrow next to the column title. By default
the table is sorted by Score.
BLAST results are calculated for the representative member of each cluster, you may see the other members (but no BLAST results) by clicking on the "Show all members" logo. Results are shown in a table with the following default columns:
UniRef100 ID
It refers to the cluster identifier. Below this there is a link to the UniRef100 report with information on the UniRef100 cluster members, as well as links to the UniRef50 and UniRef90 clusters.
Protein AC
The
Protein AC refers to the UniProtKB accession or UniParc ID. Below these identifiers, you may choose either the iProClass or the UniParc view of the protein report, respectively.
Protein
Name
The common name given to a protein,
that identifies its function or specifies its
features.
Length
Number of amino acid
residues in the peptide or protein.
Organism Name
The genus and species of the source organism from which the sequence originated. Links to NCBI taxonomy information are provided.
BLAST
Sequence Similarity Columns
There are three
columns related to BLAST results. The
Alignment column, in the extreme right, shows the
BLAST results in graphical format. The top
bar represents the query sequence. The bars below
show the region(s) on other sequences matched to
the query sequence. The bar color indicates the
magnitude of the BLAST score. Click on one of
these bars to see its BLASTalignment paired
with the query sequence.
SSearch Columns
SSearch is
a pairwise implementation of the Smith-Waterman
alignment algorithm. When two sequences are
aligned, only the shared region is shown. Within
the shared region, amino acid residues from one or
both sequences can be aligned with either amino
acids or gaps from the other sequence. The total
length of the shared region, including gaps, is
represented under the Overlap column. The
percentage of identical residues in the alignment
is given under %iden. Clicking on that number
displays the SSearch full-length
alignment.
5- Show All Members
By clicking this logo you will be able to see all sequences that belong to each cluster, though you will not see any BLAST-associated data.
FASTA Search Help
What is FASTA?
FASTA can be
used to search sequence databases, evaluate
similarity scores, and identify periodic
structures based on local sequence similarity. The
FASTA program can compare a protein sequence to a
DNA sequence database by translating the DNA
database as it is searched. This search engine
displays FASTA results (up to 200 matches), using
the FASTA program (Pearson and
Lipman, 1988) with default settings.
Select a Database
Two databases are available to perform FASTA searches: UniProtKB is the central hub for the collection of functional information on proteins, with accurate, consistent, and rich annotation. It consists of two sections: a section containing manually-annotated records with information extracted from literature and curator-evaluated computational analysis (UniProtKB/Swiss-Prot), and a section with computationally analyzed records that await full manual annotation (UniProtKB/TrEMBL).
UniRef100 provides clustered sets of sequences at 100 % from UniProt Knowledgebase (including splice variants and isoforms) and selected UniParc records (mainly from PDB, Refseq and Ensembl databases), in order to obtain complete coverage of sequence space while hiding redundant sequences (but not their descriptions) from view. It combines identical sequences and sub-fragments with 11 or more residues (from any organism) into a single UniRef entry, displaying the sequence of a representative protein, the accession numbers of all the merged UniProtKB entries, and links to the corresponding UniProtKB and UniParc records.
FASTA Search Format
The FASTA 'Search' input area accepts:
">" followed by a UniProtKB
sequence identifier. Spaces between letters
in the input are not allowed, although spaces
before or after the identifier are allowed.
Raw amino acid sequence format:MSEPQRLFFAIDLPAEIREQIIHWRATHFPPEAGRPVAADNLHLTLAFLGEVS
AEKEKALSLLAGRIRQPGFTLTLDDAGQWLRSRVVWLGMRQPPRGLIQLAN
MLRSQAARSGCFQSNRPFHPHITLLRDASEAVTIPPPGFNWSYAVTEFTLYA
SSFARGRTRYTPLKRWALTQ
The query sequence intersperse with numbers and/or spaces, such as:
1 msepqrlffa idlpaeireq iihwrathfp peagrpvaad nlhltlaflg evsaekekal
61 sllagrirqp gftltlddag qwlrsrvvwl gmrqpprgli qlanmlrsqa arsgcfqsnr
121 pfhphitllr daseavtipp pgfnwsyavt eftlyassfa rgrtrytplk rwaltq
Sequences are expected to be represented in
the standard IUB/IUPAC amino acid codes, with
these exceptions: lower-case letters are accepted
and are mapped into upper-case; U and * are
acceptable letters (see table)
Numerical digits in the query sequence are
automatically removed.
FASTA Options
-Expect
The
Expect (E-value) threshold establishes a
statistical significance threshold for reporting
database sequence matches. The default value is
0.0001, meaning that 0.0001 matches are expected
to be found merely by chance. Lower Expect
thresholds are more stringent, leading to fewer
chance matches being reported. Increasing the
expected threshold shows less stringent matches
and is recommended when performing searches with
short sequences as a short query is more likely to
occur by chance in the database than a longer one,
so even a perfect match (no gaps) can have low
statistical significance and may not be reported.
Increasing the Expect threshold allows you to look
farther down in the hit list and see matches that
would normally be discarded because of low
statistical significance.
References
Pearson WR, and
DJ Lipman (1988) Improved tools for biological
sequence comparison. Proc. Natl. Acad. Sci. USA
85(8): 2444-2448.
FASTA Results Help
A sample output for a BLAST search against the UniProtKB database is shown below

1- Query Sequence on
Click on this button to display the query sequence. Conversely, click it again to hide it.
2- Save Results As
Search results can be saved to the user's local computer. The results will be saved for selected entries or, if no proteins are selected, for all entries. Clicking "Table" will save the displayed columns as a tab-delimited text file, which may be imported into a spreadsheet for easier viewing or analysis. Clicking "FASTA" will save the IDs and sequences in FASTA format.
3- BLAST,
FASTA, Pattern Match, Multiple Alignment and
Domain Display
Retrieved entries can be
further analyzed using the sequence analysis
programs available in the Results page. First,
select the protein(s) using the checkboxes on the
left side of the table, then click the
corresponding analysis tool. Click "BLAST" or
"FASTA" button, and a new query page will be
displayed, along with the parameters that were
selected in the initial search. Click "Pattern
Match" to search against the PROSITE database. For
multiple alignment, check at least 2 proteins (but
no more than 50), then click the "Multiple
Alignment" button. A ClustalW alignment
and neighbor-joining tree will be generated along with a link to the interactive PIR viewer.
Domain display option, shows PFam domains (if
present) in graphical format.
4- Results
Display
Sort Columns
Columns can be
sorted by the corresponding values by clicking on
the arrow next to the column title. By default
the table is sorted by Score.
The results from the search are displayed
in a table with the following default columns:
Protein AC/ID
The
Protein AC/ID refers to the UniProtKB accession number and ID, respectively. Below these identifiers, you may choose either the iProClass or the UniProtKB view of the protein report. The source of the UniProtKB sequence is shown as UniProtKB/Swiss-Prot or UniProtKB/TrEMBL if the protein sequence is from the Swiss-Prot or TrEMBL section of UniProtKB, respectively.
Protein
Name
The common name given to a protein,
that identifies its function or specifies its
features.
Length
Number of amino acid
residues in the peptide or protein.
Organism Name
The genus and species of the source organism from which the sequence originated. Links to NCBI taxonomy information are provided.
PIRSF ID
If a protein belongs to a PIRSF family, then this column will display the corresponding family identifier. Click on the ID to retrieve the PIRSF report (see annotated output).
Sequence Similarity Columns
There are three
columns related to FASTA results. The
Alignment column, in the extreme right, shows the
FASTA results in graphical format. The top
bar represents the query sequence. The bars below
show the region(s) on other sequences matched to
the query sequence. The bar color indicates the
magnitude of the FASTA score. Click on one of
these bars to see its FASTA alignment paired
with the query sequence.
Related Sequence HelpRetrieve
related sequences based on pre-computed BLAST
results, therefore you will be able to have a glance at proteins similar to your query, significantly faster than running BLAST. This procedure is performed approximately every three months. Enter the UniProtKB
sequence identifier and click "Search" button.
Sample output for UniProtKB P53039
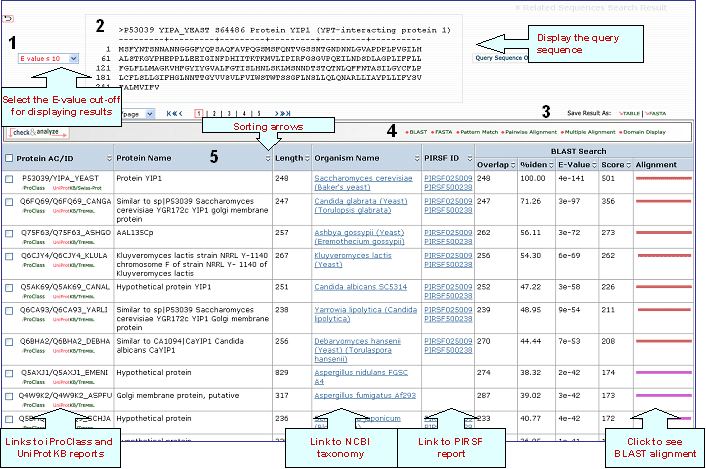
1- E-value display
Select the E-value cut-off to display your results.
2- Query Sequence on
Click on this button to display the query sequence. Conversely, click it again to hide it.
3- Save Results As
Search results can be saved to the user's local computer. The results will be saved for selected entries or, if no proteins are selected, for all entries. Clicking "Table" will save the displayed columns as a tab-delimited text file, which may be imported into a spreadsheet for easier viewing or analysis. Clicking "FASTA" will save the IDs and sequences in FASTA format.
4- BLAST,
FASTA, Pattern Match, Multiple Alignment and
Domain Display
Retrieved entries can be
further analyzed using the sequence analysis
programs available in the Results page. First,
select the protein(s) using the checkboxes on the
left side of the table, then click the
corresponding analysis tool. Click "BLAST" or
"FASTA" button, and a new query page will be
displayed, along with the parameters that were
selected in the initial search. Click "Pattern
Match" to search against the PROSITE database. For
multiple alignment, check at least 2 proteins (but
no more than 50), then click the "Multiple
Alignment" button. A ClustalW alignment
and neighbor-joining tree will be generated along with a link to the interactive PIR viewer.
Domain display option, shows PFam domains (if
present) in graphical format.
5- Results
Display
Sort Columns
Columns can be
sorted by the corresponding values by clicking on
the arrow next to the column title. By default
the table is sorted by Score.
The results from the search are displayed
in a table with the following default columns:
Protein AC/ID
The
Protein AC/ID refers to the UniProtKB accession number and ID, respectively. Below these identifiers, you may choose either the iProClass or the UniProtKB view of the protein report. The source of the UniProtKB sequence is shown as UniProtKB/Swiss-Prot or UniProtKB/TrEMBL if the protein sequence is from the Swiss-Prot or TrEMBL section of UniProtKB, respectively.
Protein
Name
The common name given to a protein,
that identifies its function or specifies its
features.
Length
Number of amino acid
residues in the peptide or protein.
Organism Name
The genus and species of the source organism from which the sequence originated. Links to NCBI taxonomy information are provided.
PIRSF ID
If a protein belongs to a PIRSF family, then this column will display the corresponding family identifier. Click on the ID to retrieve the PIRSF report (see annotated output).
Sequence Similarity Columns
There are three
columns related to BLAST results. The
Alignment column, in the extreme right, shows the
BLAST results in graphical format. The top
bar represents the query sequence. The bars below
show the region(s) on other sequences matched to
the query sequence. The bar color indicates the
magnitude of the BLAST score. Click on one of
these bars to see its alignment paired
with the query sequence.
Peptide Match
Find an exact match for a peptide sequence query in the selected database.
You can perform the search in any of these two databases: UniProtKB is the central hub for the collection of functional information on proteins, with accurate, consistent, and rich annotation. It consists of two sections: a section containing manually-annotated records with information extracted from literature and curator-evaluated computational analysis (UniProtKB/Swiss-Prot), and a section with computationally analyzed records that await full manual annotation (UniProtKB/TrEMBL).
UniRef100 provides clustered sets of sequences at 100 % from UniProt Knowledgebase (including splice variants and isoforms) and selected UniParc records (mainly from PDB, Refseq and Ensembl databases), in order to obtain complete coverage of sequence space while hiding redundant sequences (but not their descriptions) from view. It combines identical sequences and sub-fragments with 11 or more residues (from any organism) into a single UniRef entry, displaying the sequence of a representative protein, the accession numbers of all the merged UniProtKB entries, and links to the corresponding UniProtKB and UniParc records.
The peptide sequence should be in single letter code, not exceeding 30 residues long.
Peptide Match Result page
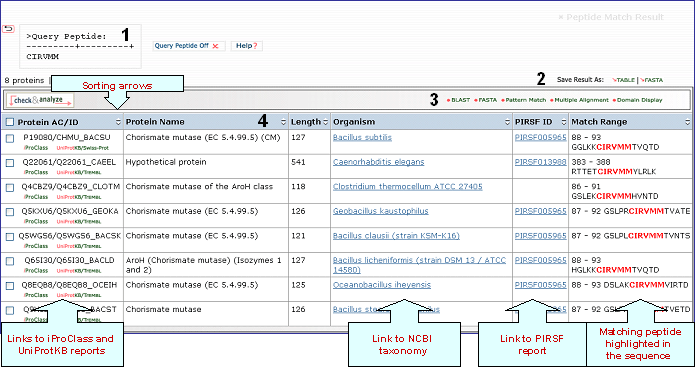
1- Query Peptide
Display ON/OFF the query peptide by clicking this box.
2- Save Results
As
The output can be saved to the
user's local computer. The results will be saved
for selected entries or, if no proteins are
selected, for all entries. Clicking "Table" will
save the displayed columns as a tab-delimited text
file, which may be imported into a spreadsheet for
easier viewing or analysis. Clicking "FASTA" will
save the IDs and sequences in FASTA
format.
3- Analyze: BLAST,
FASTA, Pattern Match, Multiple Alignment and
Domain Display
Retrieved entries can be
further analyzed using the sequence analysis
programs available in the Results page. First,
select the protein(s) using the checkboxes on the
left side of the table, then click the
corresponding analysis tool. Click "BLAST" or
"FASTA" button, and a new query page will be
displayed, along with the parameters that were
selected in the initial search. Click "Pattern
Match" to search against the PROSITE database. For
multiple alignment, check at least 2 proteins (but
no more than 50), then click the "Multiple
Alignment" button. A ClustalW alignment
and neighbor-joining tree will be generated along with a link to the interactive PIR viewer.
Domain display option, shows PFam domains (if
present) in graphical format.
4- Results Display
Results of
the search are displayed in a table.
Protein AC/ID
The
Protein AC/ID refers to the UniProtKB or UniRef100 identifiers. Below these numbers, you may choose either the iProClass or the UniProtKB/UniRef100 view of each protein report. The source of the UniProtKB sequence is shown as UniProtKB/Swiss-Prot or UniProtKB/TrEMBL if the protein sequence is from Swiss-Prot or TrEMBL, respectively.
Protein Name
The common
name given to a protein, that identifies its
function or specifies its features.
Length
Number of amino acid
residues in the peptide or protein.
Organism Name
The genus and species of the source organism from which the sequence originated. Links to NCBI taxonomy information are provided.
PIRSF ID
If a protein belongs to a PIRSF family, then this column will display the corresponding family identifier. Click on the ID to retrieve the PIRSF report (see annotated output).
Match
Range
This column displays in red the peptide query within the sequence.
Pattern Search
Find a PROSITE pattern in
a query sequence, entering the single amino acid code sequence or its
unique ID, and search against the PROSITE database
or, alternatively, you may define a pattern
and search against a selected sequence database. A
pattern is a formula (regular expression) that
represents the conserved region of a group of
related proteins. Once constructed, the pattern is
used by a pattern-matching program to find
possible occurrences of the conserved region in
the sequence database.
How to write a protein
pattern
Follow the example below to build
the protein pattern.
[LIVM]-[VIC]-x(2) -G-[DENQTA]-x-[GAC]-x(2) -[LIVMFY](4)-x(2)-G
- Use capital letters for amino acid residues
and put a "-" between two amino acids (not
required).
- Use "[�] for a choice of
multiple amino acids in a particular position.
- [LIVM] means that L, I,
V, or M can be in the first
position.
- Use "{�}" to exclude amino acids.
- {CF} means C and F should not be in that
particular position
- Use "x" or "X" for a
position that can be any amino acid.
- Use "(n)", where
n is a number, for multiple positions.
- x (3) is the same as "xxx"
- Use "(n1,n2)" for multiple or variable
positions.
- " x (1,4) represents "x" or "xx" or "xxx" or
"xxxx"
- Use the symbol ">" at the beginning or
end of the pattern to require the pattern to
match the N or C terminus.
- ">MDEL" finds only sequences that start
with MDEL
- "DEL>" finds only sequences that end with
DEL
[LIVM]-[VIC]-x(2) -G-[DENQTA]-x-[GAC]-x(2) -[LIVMFY](4)-x(2)-G Illustrates a 17 amino acid peptide that
has: an L, I, V, or M at position 1; a V, I, or C
at position 2; any residue at positions 3 and 4; a
G at position 5 and so on �.
Note: You can
also write the above pattern as:
[LIVM] [VIC] X
(2) G [DENQTA] X [GAC] X (2) [LIVMFY] (4) X (2)
G
Result for proteins with PROSITE pattern PS00888 (Cyclic nucleotide-binding domain signature 1)

1- Save Results
As
The output can be saved to the
user's local computer. The results will be saved
for selected entries or, if no proteins are
selected, for all entries. Clicking "Table" will
save the displayed columns as a tab-delimited text
file, which may be imported into a spreadsheet for
easier viewing or analysis. Clicking "FASTA" will
save the IDs and sequences in FASTA
format.
2- BLAST,
FASTA, Pattern Match, Multiple Alignment and
Domain Display
Retrieved entries can be
further analyzed using the sequence analysis
programs available in the Results page. First,
select the protein(s) using the checkboxes on the
left side of the table, then click the
corresponding analysis tool. Click "BLAST" or
"FASTA" button, and a new query page will be
displayed, along with the parameters that were
selected in the initial search. Click "Pattern
Match" to search against the PROSITE database. For
multiple alignment, check at least 2 proteins (but
no more than 50), then click the "Multiple
Alignment" button. A ClustalW alignment
and neighbor-joining tree will be generated along with a link to the interactive PIR viewer.
Domain display option, shows PFam domains (if
present) in graphical format.
3- Results Display
Results of
the search are displayed in a table. Columns can be
sorted by the corresponding values by clicking on
the arrow next to the column title.
Protein AC/ID
The
Protein AC/ID refers to the UniProtKB accesion and identifier, respectively. Below these, you may choose either the iProClass or the UniProtKB view of each protein report. The source of the UniProtKB sequence is shown as UniProtKB/Swiss-Prot or UniProtKB/TrEMBL if the protein sequence is from Swiss-Prot or TrEMBL, respectively.
Protein Name
The common
name given to a protein, that identifies its
function or specifies its features.
Length
Number of amino acid
residues in the peptide or protein.
Organism Name
The genus and species of the source organism from which the sequence originated. Links to NCBI taxonomy information are provided.
PIRSF ID
If a protein belongs to a PIRSF family, then this column will display the corresponding family identifier. Click on the ID to retrieve the PIRSF report (see annotated output).
Match
Range
This column displays the sequence range for the query pattern.
Partial result for PROSITE patterns in UniProtKB O05689

Multiple Alignment Help
Enter multiple sequences with
their corresponding ID lines in FASTA
format or enter multiple UniProtKB
IDs separated by lines, commas or spaces. Then
click the "Multiple Alignment" button, and the
ClustalW multiple alignment (CLUSTALW v1.8,
Thompson et al., 1994), neighbor-joining tree and a PIR interactive tree and alignment viewer
will be generated. See sample output
FASTA Format
A sequence in FASTA format begins with a single-line description, followed by lines of sequence data. The description line is distinguished from the sequence data by a greater-than (">") symbol in the first column. One benefit of using FASTA format is that the sequence identifier will be reported with the results. An example sequence in FASTA format is:
>gi|3287971|sp|P37025|LIGT_ECOLI 2'-5' RNA ligase
MSEPQRLFFAIDLPAEIREQIIHWRATHFPPEAGRPVAADNLHLTLAFLGEVSAEK
EKALSLLAGRIRQPGFTLTLDDAGQWLRSRVVWLGMRQPPRGLIQLANMLRSQA
ARSGCFQSNRPFHPHITLLRDASEAVTIPPPGFNWSYAVTEFTLYASSFARGRTRY
TPLKRWALTQ
References
Thompson JD,
Higgins DG, and TJ Gibson (1994) CLUSTAL W:
improving the sensitivity of progressive multiple
sequence alignment through sequence weighting,
position-specific gap penalties and weight matrix
choice. Nucleic Acid Res.
22(22):4673-4680.
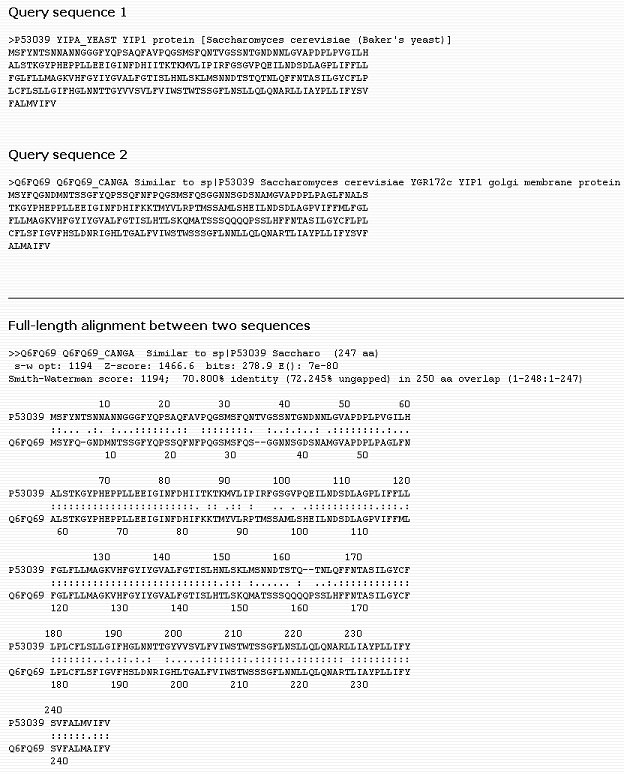
Pairwise Alignment Help
Insert two sequences using the
single letter
amino
acid code or enter two UniProtKB identifiers codes. Press the "Submit" button
and the alignment results will show the SSEARCH
Smith-Waterman full-length alignments between two
sequences (SSEARCH program version 3.4t24 July 21, 2004).
References
Smith TF, and MS Waterman (1981) Identification of common
molecular subsequences. J. Mol. Biol. 147,
195-197.
Pairwise alignment of P53039 and Q6FQ69
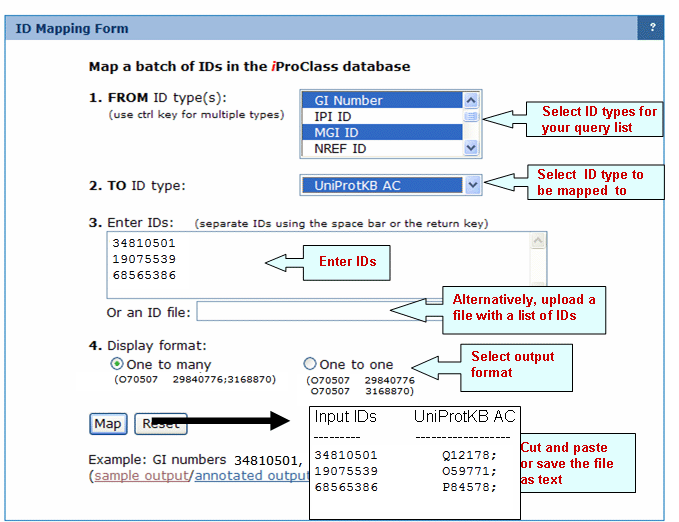
ID Mapping Help
This function allows to map a batch of IDs in the iProClass database.
First specify the type of ID of your query, then specify the type of ID you want to see the correspondence to. Type the identifiers in the box provided separating them by a space or return key, and press "Go Map".
In the result page, click on "show match list" to see the table of correspondence between the selected IDs. You can cut and paste the identifiers for other purpose if needed.
In addition, you will get a table similar to batch retrieval where you will be able to further analyzed your sequences. See annotated output below.
Mapping of GI numbers to UniProt ACs
Composition/Molecular Weight Calculation
Help
Retrieve the composition
information and the molecular weight of a sequence
based on the UniProtKB identifier or the single letter amino
acid code. The results contain the number of
residues and percentage for each amino acid in the
sequence. The calculation notes for the molecular
weight and the molecular weight for each amino
acid are also given.
Molecular weight composition for P53039
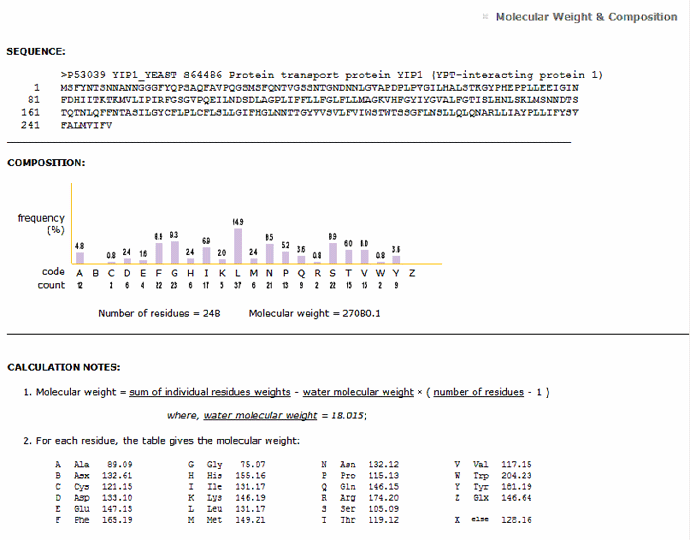
PIRSF
Scan
Search your
query sequence against the fully curated PIRSF
families. When you submit your query sequence, it
will be searched against the full-length and
domain HMM models for the fully curated PIRSFs by
HMMER program. If a match is found, the matched
regions and statistics will be displayed. The
query sequence should be entered using single
letter code or a UniProtKB identifier.
PIRSF scan for Q8Y5X7

Amino Acid Sequence Code Table
| Code |
Amino Acid |
| A | alanine |
| B | aspartate or asparagine |
| C | cysteine |
| D | aspartate |
| E | glutamate |
| F | phenylalanine |
| G | glycine |
| H | histidine |
| I | isoleucine |
| K | lysine |
| L | leucine |
| M | methionine |
| N | asparagine |
| P | proline |
| Q | glutamine |
| R | arginine |
| S | serine |
| T | threonine |
| U | selenocysteine |
| V | valine |
| W | tryptophan |
| Y | tyrosine |
| BLAST and FASTA also accept |
| X | any |
| * | translation stop |
UniProtKB Identifiers for BLAST/FASTA
Search and Analysis Tools
The
following table shows examples for UniProtKB accession number and ID
|
| iProClass Report |
 |
|
| UniProtKB Report |
 |
|
| UniParc Report |
 |
|
| PIRSF Report |
| Sample report for PIRSF000186. Information about the numbered fields is provided below. |
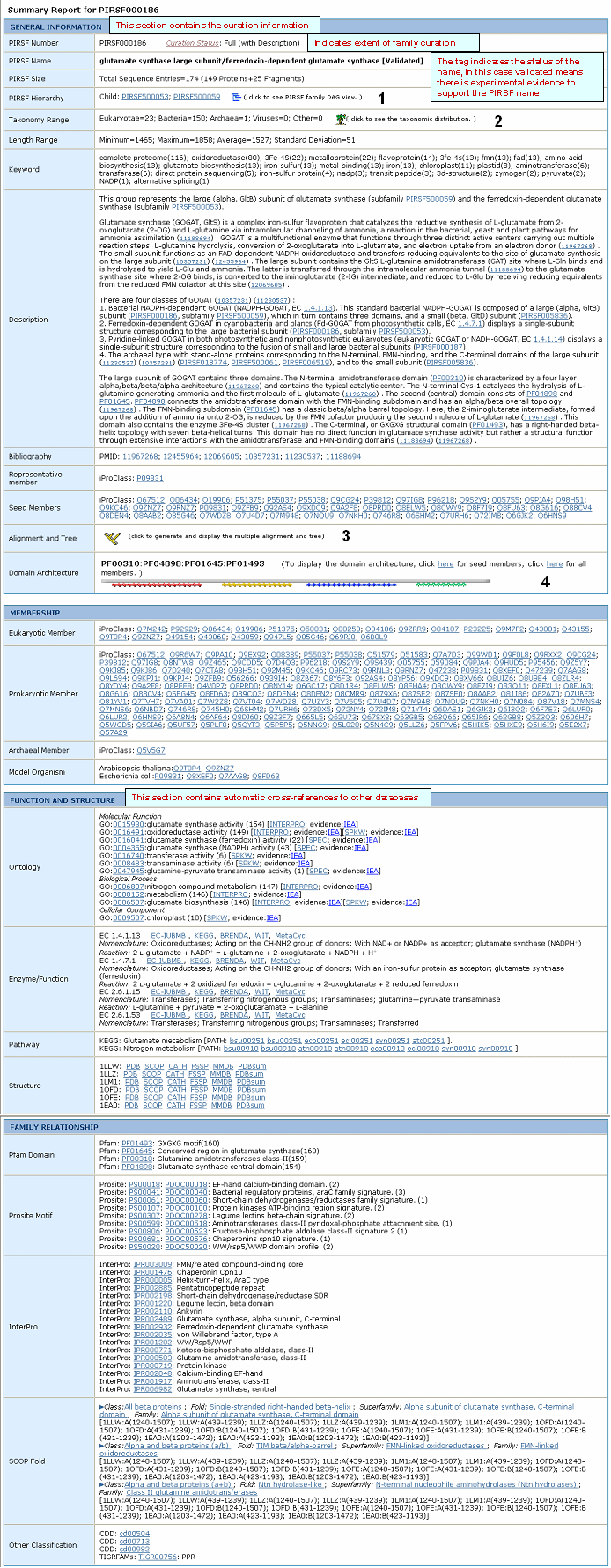 |
| 1- PIRSF Hierarchy DAG View |
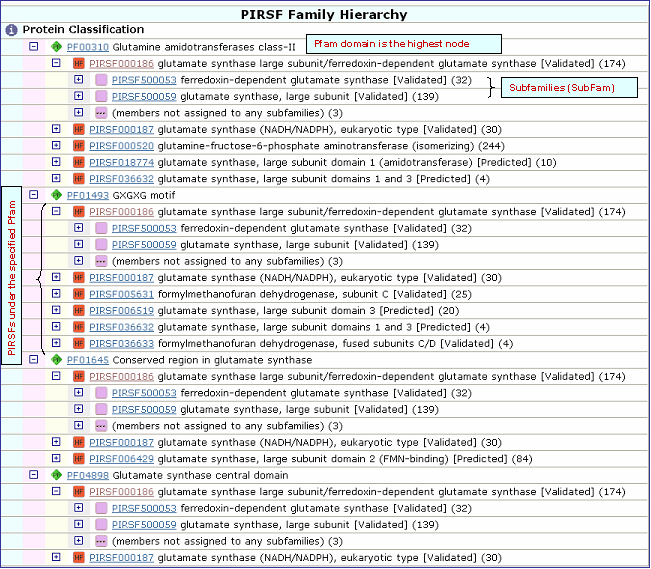 |
| 2- PIRSF Taxonomic Distribution |
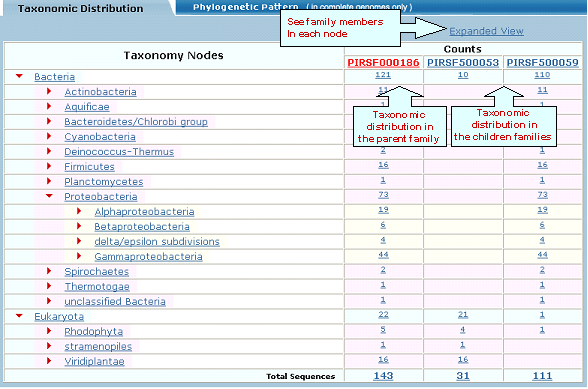 |
|
3- Multiple Alignment |
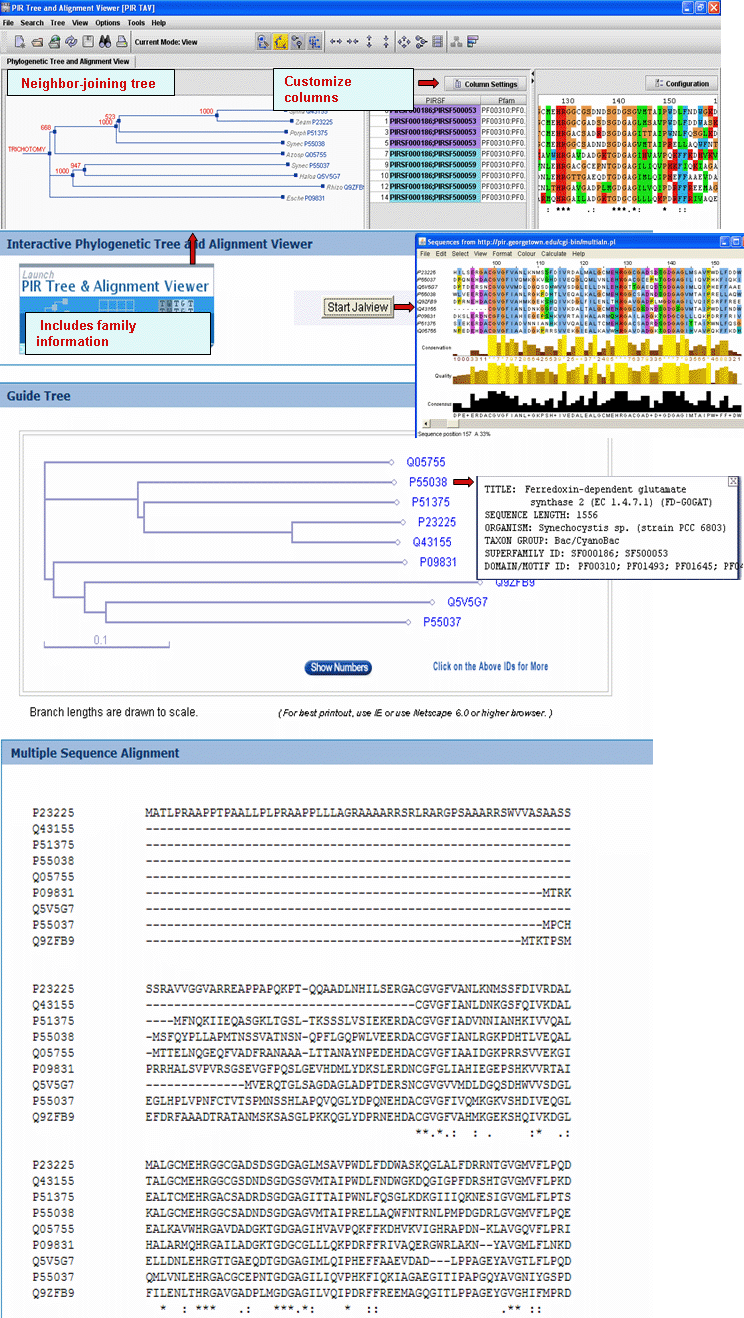 |
| 4- Domain Architecture |
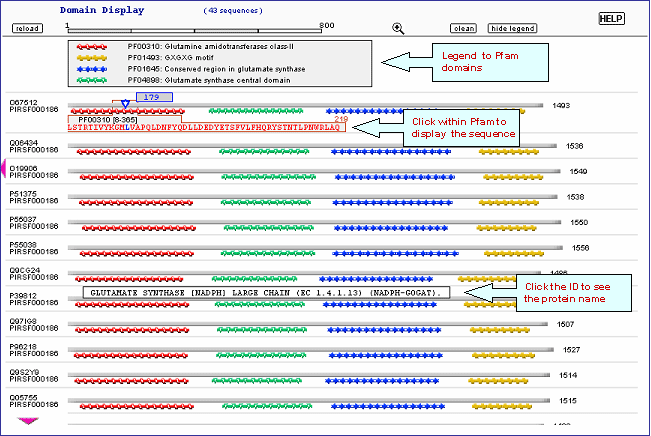 |
|
|


